04/18/23
A Primer on Preclinical Toxicologic Pathology
notes on Investigative New Drug enabling studies
R&D
R&D refers to the pharmaceutical research and development of new medicines.
R is research. Research in the drug life cycle is mostly drug discovery and bit of preclinical.
D is development. Development in the drug life cycle is preclinical and clinical trials. The purpose of drug development is to determine the efficacy (benefit) and safety (risk) as tied to the body’s exposure to the drug (pharmacokinetic profile).

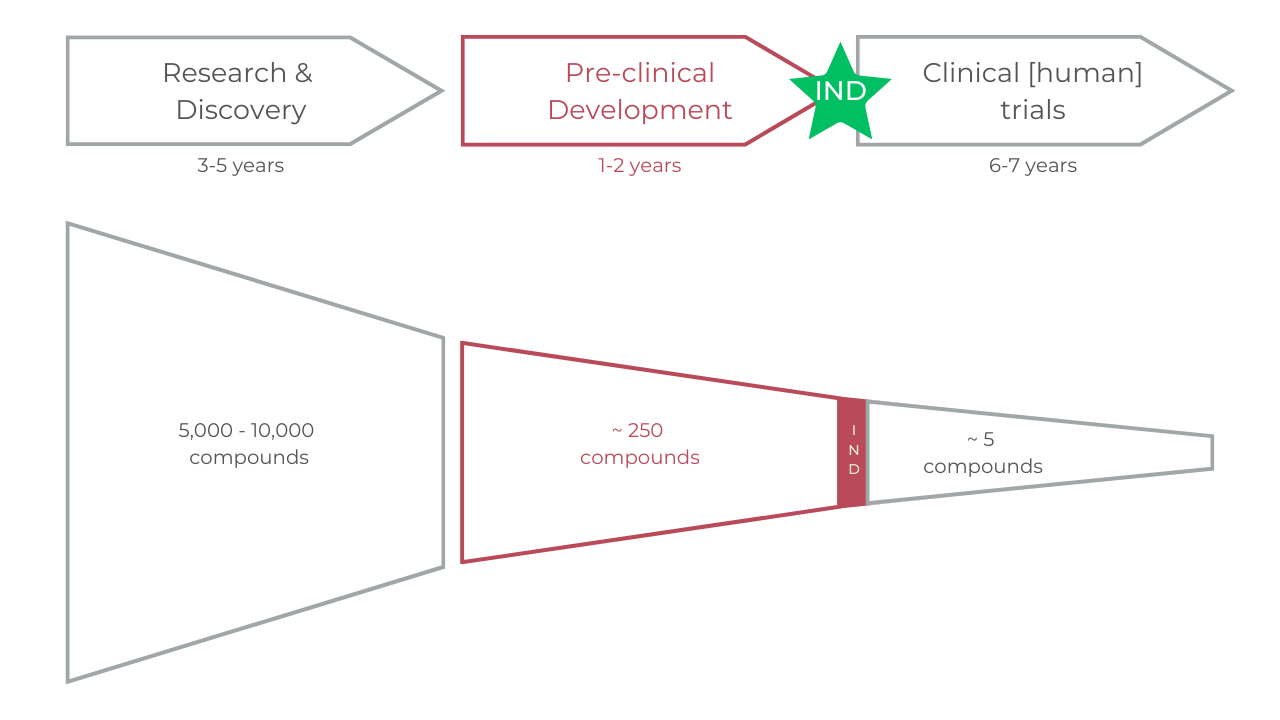
The process is three fold
Research & Discovery – during this stage, scientists want to understand the disease and select a target (usually a receptor site on a cell) that can potentially be affected by a drug molecule.
This can involve screening large numbers of compounds for potential efficacy and identifying potential targets for treatment.
Preclinical Development – during this stage, scientists conduct extensive testing on cell cultures and animal models to evaluate the safety and efficacy of the drug candidate.
This stage includes toxicology and pharmacokinetics studies. This primer is most interested in this stage.
Clinical Trials – after preclinical testing, if the FDA issues an Investigational New Drug (IND), the drug candidate moves on to clinical trials.
These trials are conducted in humans to evaluate safety and efficacy.
The IND – Investigational New Drug
Celine Halioua has done a great primer on the IND. Much of the below is lifted from her work. I’ve added toxpath-related details for consideration.
The federal definition of a drug is defined as
a product used in diagnosing, curing, mitigating, treating or preventing a disease or affecting a structure or function of the body.
Before you can start Phase 1 clinical trials with an experimental medicine, you must file an IND with the FDA. You need to submit an IND if you are testing a New Molecular Entity, or a known drug for a new indication.
The IND (Investigative New Drug) application is required to test in humans.
The purpose of an IND is threefold: Firstly, it allows for the interstate transport of non-market approved drugs. Secondly, it grants the FDA authority to regulate experimental medicines. Finally, it permits the agency to oversee the safety of the experimental compound, ensuring that patients and volunteers are not exposed to unreasonable risks.
For drug development and biotech startups, the most relevant type of IND is the investigator IND, which is used for human testing of a new drug or new use. Additionally, an emergency use IND exists for experimental drugs used outside of standard clinical trials.
After submitting an IND, the sponsor must wait for 30 calendar days before beginning any clinical trials. During this time, the FDA reviews the IND for safety to ensure that research subjects are not exposed to unreasonable risks. Once this period has elapsed, the IND is considered "open," and the investigator is responsible for updating it through amendments. These amendments can include protocol amendments, informational amendments (such as those related to toxicology or chemistry), safety reports (covering adverse events in clinical trials or ongoing laboratory animal studies), and annual reports.
What is ToxPath?

Toxicologic pathology is a medical discipline that applies the professional practice of pathology—the study of diseases—to toxicology—the study of the effects of chemicals and other agents on humans, animals, and the environment.
Both toxicology and pathology are critical components of the safety assessment process in preclinical development used in predicting human and animal responses to drugs, chemicals, and therapeutic devices, including identifying the potential of these agents to cause cancer.
In addition to morphological diagnoses of organ and tissue pathology, toxicologic pathology includes identifying injury at the cellular, subcellular, and molecular levels to identify the health consequences of the adverse effects of drugs, chemicals, and therapeutic devices on aging, genetics, nutrition, immune function, developmental biology, reproduction, and brain development.
Why is ToxPath important?
Preclinical toxicology studies are critical to identify potential safety issues before the drug is tested in humans. It is a very important step to determine the safe dosage and side effects for human use.
ToxPath is the bottleneck for the safety of any drug candidate ever.
As you will see below, toxpath is an important part of the IND process.
Investigative New Drug (IND) Application Flow
The IND can be divided into three main sections:
Animal pharmacology and toxicology studies, which include preclinical data that confirms the experimental drug's safety for human use, guidance for dosing and rationale, any previous use in humans (if applicable), potential risks to patients caused by the drug's chemistry, and an assessment demonstrating the ability to manufacture the drug in large enough batches consistently.
Manufacturing information, which provides details on the drug's structure, composition, and final formulation, along with relevant data such as stability and manufacturer information.
Clinical protocols, which outline protocols for the clinical studies, the relevance of these protocols to the drug's proposed safety and efficacy, the clinical investigator's qualifications, and the institutional review board (IRB) and informed consent.
Once submitted, the FDA has 30 days to review the IND and raise any objections. Deficiencies and required clinical holds may include unreasonable risk of harm to human subjects, unqualified investigators, inadequate protocol design, disagreements over starting dosage, inadequate CMC information, a misleading, incomplete, or erroneous Investigator's Brochure, gender bias in a study of patients with a life-threatening disease that affects both genders. Preclinical safety PK/PD considerations include dose formulation analysis, genomic toxicity, mutagenicity, chromosomal aberrations, range-finding for tox, acute tox, metabolism, hERG test, cardiovascular, respiratory, and CNS safety.
IND Table of Contents
The IND itself roughly takes the following form:
Form FDA 1571
Table of Contents
Introductory Statement
General Investigation Plan
Investigator’s Brochure
Protocol(s) Phase I
Study Protocol(s)
Outline of study
Estimate of # of patients
Safety exclusions
Dosing
Monitoring for AEs
Investigator Data
Facilities Data
Institutional Review Board (IRB) or Completed Forms
Chemistry, Manufacturing, and Control Data
Drug substance
Drug product
Placebo
Labeling
Environmental Assessment or Claim for Exclusion
(Most submissions will qualify for exclusion)
Pharmacology and Toxicology Data
Pharmacology summary
Pharmacokinetics
Toxicology summary
Qualifications of those who evaluated the safety data
Previous Human Experience
Additional Information
GMP & GLP – Good Manufacturing and Good Laboratory Practices
During preclinical development, if scientists start considering testing or using a compound in humans, generally speaking GLP regulations apply.
Good Laboratory Practice is a set of quality management controls for research laboratories. It is a data quality system.
Good Manufacturing Practice is a system for ensuring consistency in quality for manufacturing. It covers from starting materials to staff training.
Work conducted under GLP/GMP conditions is generally more expensive and complicated.
Animal Toxicology Studies in the IND Package
Preclinical studies are conducted to establish evidence of a drug's safety and ensure that it does not pose undue risk to patients or subjects. It is crucial to carry out preclinical studies across mammalian species to avoid unintended bias or oversight of important drug characteristics, particularly those related to safety.
Effective planning is essential, as many of these studies are interrelated. The toxicology studies in the IND package include acute, subacute, and chronic toxicity tests, effects on reproduction and the developing fetus, toxicity relevant to the modality and use case, target organ toxicity, local tolerance (related to the mode of administration), in vitro studies, toxicokinetics, carcinogenic potential, and genotoxicity studies. Depending on the drug's characteristics, these studies may also include assessments of phototoxicity, immunotoxicity, juvenile animal toxicity, and abuse liability.
For drugs intended to treat severe diseases, modifications are made to the standard preclinical package to streamline and accelerate the drug development process.
Toxicity Study Types
Toxicity studies are usually done after pharmacology (PK/PD) studies, and are conducted using the same route as human delivery. Importantly, the duration of the clinical trial is limited by the duration of preclinical toxicity repeated dose studies. Toxicity studies should be conducted at low (no effect), mid (near anticipated human dose), and high (10-100x higher than anticipated human clinical dose).
In preclinical, the following toxicity studies are administered
Acute toxicity studies
Repeated-dose toxicity
Dosing
Estimating the first dose in humans
Clinical Protocol
The IND will include a General Investigational Plan, which gives an overview of:
The drug rationale
Indication
How the drug will be evaluated
The type of clinical trials
Number of patients in these clinical trials
Particular risks to be considered as informed by preclinical data or prior human studies
Market Segmentation
Toxicologic pathology professionals work in academic institutions, government, the pharmaceutical and chemical industry, CROs or as consultants, and utilize traditional clinical or anatomic pathology endpoints, as well as contemporary advances in molecular and cellular biology. They are dedicated to the integration of toxicologic pathology into hazard identification, risk assessment, and risk communication regarding human, animal, and environmental exposure to potentially toxic substances.
By End User
By end user, the market includes pharmaceutical companies, biotechnology companies, and contract research organizations (CROs). Pharma and biotech outsource most of their preclinical toxpath to CROs.
By Drug Type
By drug type, the market includes small molecule drugs and biologics drugs.
By Therapeutic Area
By therapeutic area, the market includes oncology, cardiovascular diseases, respiratory diseases, and diabetes.
The Cost of Science
As a starting point, here is a sample study run by a CRO in animal models and its cost. via Celine Halioua.
Facilities
Vivarium, 100 mice, shared room - $4,000/month
Bench and desk in a shared lab space - $1,000/month
Technician cost - $150+/hour
Experiments
Testing four compounds in a cell line - $10,000
Cell line gene knockdown, with materials and assays - $45,000
Mouse study, n=15, 6 week drug dosing with assays - $30,000
Mouse study, drug dosing, functional readouts - $130,000
Old mouse lifespan study, with drug, minimal assays - $65,000
Young mouse lifespan study, with drug, functional and biomarker assays - $250,000
Large animal, small n, 30d study + assays - $50,000
Large animal, large n, 30d study + assays - $400,000
Target Animal Safety - $400,000 - $1M
Assays
These numbers are at moderate scale - 100-400 samples run. This can become a lot more expensive if you only run a few samples.
Assay development (ELISA) - $30,000
Running the optimized ELISA - $50/each
Clinical chemistry on serum - $50-155/sample - you usually have to pay on top of this to have the data interpreted
Read more about common costs in preclinical testing programs here.
This primer is 18/50 of my 50 days of learning. Subscribe to hear about new posts.